Another anti-TREM2 antibody looks like a promising candidate for treating Alzheimer’s disease. In the June 24 Journal of Experimental Medicine, researchers led by Marco Colonna at Washington University, St. Louis, and Tina Schwabe, Alector LLC, South San Francisco, report that the biotech company’s AL002c activates TREM2 in a mouse model of amyloidosis.
- Antibody activated TREM2 signaling in mouse microglia.
- Chronic treatment reduced neurotoxicity and inflammatory signaling.
- AL002 appeared safe in Phase 1 trial, boosting sTREM2 in CSF.
While some microglia proliferated in response, the number of microglia around amyloid plaques held constant, as did overall plaque load. Even so, treated mice had fewer dystrophic neurites and they behaved more like wild-type mice in open-field tests, suggesting the treatment attenuated Aβ toxicity. It appears that in treated mice, microglia extended more processes around plaques, clearing diffuse, filamentous material while leaving the compact, presumably inert aggregates. “Thus, AL002c reduces the formation of ‘neuritic’ plaques and the associated induction of swollen, degenerating axons and dendrites,” the authors concluded. Colonna is on Alector’s scientific advisory board.
This publication comes on the heels of a paper in EMBO Molecular Medicine in March reporting the effects of another activating TREM2 antibody, 4D9. Researchers led by Christian Haass at Ludwig Maximilians University, Munich, and Joseph Lewcock at Denali Therapeutics, South San Francisco, reported that 4D9 did reduce plaque load in APP knock-in mice, mostly by gobbling up the diffuse amyloid around the plaque cores (Mar 2020 news).
Neurites Nullified. After three months of weekly antibody injections (right), mouse brain had fewer dystrophic neurites (red) surrounding Aβ plaques (blue). [Courtesy of Wang et al., JEM 2020.]
The groups disagree on how the two studies compare. Colonna considers them to be quite different; Haass sees them as being quite similar. The main point of departure is whether this therapeutic strategy reduces plaque load. That may, in part, come down to the models used. Haass and colleagues tested 4D9 in APP NL-GF mice, which tend to have more diffuse plaques than do the 5xFAD crosses Colonna and colleagues used.
For the AL002c study, co-first authors Shoutang Wang, Meer Mustafa, and colleagues crossed 5xFAD/TREM2 KO mice with mice expressing either the common (CV) or R47H variant of human TREM2 (Song et al., 2018). The R47H partial loss-of-function variant increases a person’s risk of developing AD.
When the CV and R47H crosses were 5 months old, and amyloid plaques were already established in the brain, Wang treated them weekly with 30mg/Kg of AL002c. Once the mice were 7.5 months old, the authors tested them in the elevated-plus maze, then killed them at 8 months to see how the antibody had affected the brain.
The plus maze has open and covered arms; mice tend to favor the latter. Untreated 5xFAD/hTREM2 R47H and 5xFAD/hTREM2 CV were less cautious and ventured into the open arm more often, but AL002c curtailed this abnormal behavior. “This is not a classic memory test, but it is robust in the 5xFAD model and correlates highly with pathology,” noted Colonna.
The pathology changes were mixed. Filamentous/diffuse plaque load nudged downward 10 percent in AL002c-treated mice that expressed the common variant of human TREM2, while the amount of “inert” plaques nudged up by about the same amount. Wang and colleagues saw a similar shift in the treated R47H hTREM2 mice. Filamentous plaque load dropped and inert plaque load increased.

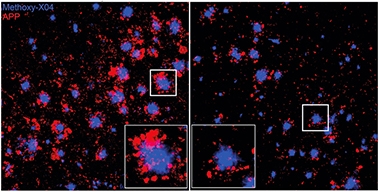
Plaque Troika. On the basis of methoxy-O4 (green) and 6E10 (red) binding, the authors identified filamentous (red arrowhead), inert (green arrowhead), and compact plaques (yellow arrowhead). [Courtesy of Wang et al., JEM 2020].
The authors define filamentous plaques as those with little β-sheet core and fibrils that extend to the parenchyma; inert plaques as those forming a compact β-sheet core with no Aβ fibrils extending into the surrounding parenchyma; and compact plaque as those with a β-sheet core and non-β sheet protrusions (see image at left). The antibody had no effect on compact plaques.
What caused the shift in plaque composition? The authors used single-cell RNA-Seq to try to catch transcriptional changes among microglia in situ after the mice had been on AL002c for two days. They isolated and phenotyped more than 40,000 CD45-positive cells. Grouping them by transcriptome similarity, they found that 12 of 20 clusters, accounting for more than three-quarters of all isolated cells, were microglia. Some had homeostatic transcriptomes, others the disease-associated type, and some were “transitional,” i.e., hybrids of both. Still others were in mitosis.
AL002c treatment brought more transitional and proliferative cells, and suppressed more homeostatic ones, than seen in untreated mice. The effect was more robust in the R47H hTREM2 than the common variant mice. Colonna thinks this is because R47H microglia are less responsive to endogenous ligands, and therefore are less active to begin with.

Probing Plaques. In mice treated with AL002c (right), microglia (red) extended more processes (rendered in light blue) around amyloid plaques (dark blue) than did microglia in mice treated with a control antibody (left). [Courtesy of Wang et al., JEM 2020.]
Though the antibody increased microglial proliferation, there were no additional microglia milling around amyloid plaques after three months of treatment. Analysis of Iba1+ cells and methoxy-O4-positive plaques in 8-month-old treated mice showed similar microglial “coverage” of hippocampal plaques in treated and untreated mice. In the cortex, there even seemed to be fewer microglia around plaques.
Instead, the change seemed to lie in microglial activity. The cells extended more feelers around the plaques (see image above right) and they more actively phagocytosed Aβ42 (see image below), and they reduced the number of dystrophic neurites.
“The surprise, which perhaps should not be much of a surprise, is we saw a decrease of microglial clustering around plaques,” Colonna said. “It seems that as we reduced the toxicity of plaques, a secondary response was a reduction of microglia. Essentially we reduced microgliosis.” In support of this, levels of the inflammatory microglial marker, Spp1, fell in treated mice.

Phagocytosed. In 5xFAD/CV hTREM2 mice treated with AL002c (bottom) more A42 (green) associated with microglial phagosomes (red) than in untreated mice (top). [Courtesy of Wang et al., JEM 2020.]
Is failure to clear plaques a shortcoming? Colonna does not think so. “We’ve seen from immunotherapy trials that clearing plaques is not necessarily beneficial,” he said. However, he cautioned that it is unclear what will happen after chronic activation of TREM2. “You could potentially overstimulate the cells, desensitize the receptor, and cause cell death or anergy,” he said.
This could be one interpretation of biomarker data from a Phase 1 trial of AL002, a clinical equivalent of AL002c. In this study of 56 healthy volunteers, analysis of cerebrospinal fluid revealed a dose-dependent decrease of the shed, soluble extracellular domain of TREM2, which could mean the antibody stimulated cellular uptake of the receptor. Equally, it could mean the antibody interfered with proteolytic cleavage of TREM2. “Whether a reduction of sTREM2 in the brain contributes to the beneficial effect of the antibody remains to be determined,” the authors wrote.
CSF levels of sCSF-1R, a shed fragment of the microglial colony-stimulating factor receptor, increased in parallel. CSF-1R is an essential receptor for microglial survival, and the sCSF-1R might reflect an increase in proliferation of the cells, the authors suggest.
The treatment seemed well-tolerated with no serious adverse events up to 12 weeks after a single dose. Alector’s Robert Paul told Alzforum that the company plans to start a Phase 2 trial by the end of this year.—Tom Fagan
News Citations
Research Models Citations
- APP NL-G-F Knock-in
- Trem2 KO (Colonna) x 5XFAD
- TREM2, humanized (common variant) X 5XFAD
- TREM2, humanized (R47H) X 5XFAD
Therapeutics Citations
Paper Citations
- Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, Colonna M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med. 2018 Mar 5;215(3):745-760. Epub 2018 Jan 10 PubMed.
External Citations
No Available Further Reading
"load" - Google News
June 27, 2020 at 07:17AM
https://ift.tt/2YBXR35
In Mice, Activating TREM2 Tempers Plaque Toxicity, not Load - Alzforum
"load" - Google News
https://ift.tt/2SURvcJ
https://ift.tt/3bWWEYd
Bagikan Berita Ini














0 Response to "In Mice, Activating TREM2 Tempers Plaque Toxicity, not Load - Alzforum"
Post a Comment